![]()
Distillazione.
Operazione di separazione dei componenti di una miscela per mezzo di una parziale evaporazione di questa e recupero separato di una fase gassosa e di un residuo liquido. Essa si basa sulla diversa volatilità (cioè tendenza ad evaporare) che hanno i diversi componenti di una miscela: quelli più volatili (o più leggeri) vengono estratti nella fase gas, mentre quelli più pesanti (cioè meno volatili) vengono recuperati nella fase liquida residua. La d. è quindi un caso particolare della vasta operazione di frazionamento di miscele. Occorre premettere che la separazione non può mai essere completa. è possibile spingerla fin che si vuole, almeno da un punto di vista teorico, cioè fino a recuperare un componente puro al 99% o 99,9% o 99,99% e così via, ma mai fino ad avere un componente puro al 100%. Per far ciò occorrerebbe un'apparecchiatura di grandezza infinita, cosa evidentemente impossibile. In pratica ogni componente della miscela è recuperato con la purezza minima indispensabile per l'uso cui è destinato. è evidente d'altra parte che una stessa sostanza deve avere una purezza ben diversa secondo la sua destinazione (ad esempio per la fabbricazione di concimi o di medicinali); naturalmente nei due casi alla diversa purezza corrispondono prezzi ben diversi. Innanzitutto è necessario chiarire la terminologia propria dell'impiantistica chimica, in modo da evitare equivoci. Citiamo alcuni esempi. è noto a tutti che da una soluzione di cloruro sodico (il comune sale da cucina) è possibile recuperare il sale contenuto evaporando tutta l'acqua.
![]()
![]()
Questa operazione viene detta propriamente evaporazione, in quanto solo uno dei componenti della miscela (l'acqua) è volatile. L'evaporazione riguarda quindi la separazione di miscele nei loro componenti volatili da quelli non volatili (generalmente solidi a fine operazione). Se la separazione non è completa si parla di concentrazione. La d. è invece l'operazione di separazione di miscele di componenti tutti volatili (alcuni più, alcuni meno). Essa si differenzia ancora dall'operazione di assorbimento e deadsorbimento o stripping in quanto la fase vapore presente è generata per riscaldamento del liquido, onde il gas ed il liquido a contatto contengono gli stessi componenti, anche se con concentrazioni diverse. Ad esempio si consideri una soluzione di ammoniaca in acqua, quale potrebbe essere la comune ammoniaca (liquida) in commercio in soluzione al 37%. Se attraverso questo liquido si fa passare una corrente d'aria (che è praticamente insolubile in acqua) questa estrae l'ammoniaca dalla soluzione, onde si è realizzata una separazione. Si tratta però di uno stripping o deadsorbimento, in quanto si ha alla fine un gas che contiene componenti (l'ossigeno e l'azoto dell'aria) che non erano presenti nella soluzione; il recupero dell'ammoniaca pura si presenta poi complesso. Se invece la stessa soluzione di ammoniaca viene riscaldata in un ambiente chiuso, la fase gassosa che si forma contiene solo ammoniaca e vapor d'acqua, ma le proporzioni di NH3 e H2O sono diverse da quelle presenti nella soluzione; precisamente il gas è molto più ricco di ammoniaca, onde si è realizzata una certa separazione. In questo caso si tratta di una d. Se si pensa di condensare completamente il gas ottenuto e poi di ripetere di nuovo sul condensato l'operazione di d., si ottiene una fase gassosa più ricca in NH3 di quella ottenuta la volta precedente; facendo quindi molte operazioni successive è possibile ottenere NH3 alla purezza voluta (ad es. 98%), le impurezze essendo sempre vapore d'acqua. Nella maggior parte dei casi questo metodo è poco usato perché lungo e costoso. Esso ha però un'applicazione nota da secoli: la produzione di acquavite dal vino o dalle vinacce. Per d. successive si ha una fase acquosa, poverissima in alcool ed una fase gassosa (o liquida, dopo condensazione) che è ricca in alcool, cioè nel componente più volatile della miscela, e costituisce la grappa distillata. Insieme all'alcool vengono distillate anche altre sostanze molto volatili, che conferiscono al prodotto il caratteristico odore e sapore. Nella maggior parte dei casi la d. è accoppiata alla rettifica. L'operazione di rettifica è ancora un'operazione di separazione di miscele: consiste nella condensazione parziale di una miscela gassosa. Si ha un fenomeno parallelo a quanto avviene nella d.: se la condensazione è parziale si ottiene una fase liquida ed una gassosa, ma le concentrazioni dei diversi componenti non sono uguali nelle due fasi. Precisamente: la fase gassosa è più ricca nel componente più volatile della miscela di partenza, mentre la fase liquida ne è più povera (cioè è più ricca nel componente più pesante). Lo stesso discorso vale naturalmente per miscele a più di due componenti, salvo che i calcoli sono più complessi. Viene quindi naturale pensare ad un accoppiamento di queste due operazioni, cioè la realizzazione di quanto viene detto d. con rettifica. In questo caso viene sfruttata la separazione parziale realizzata sia con la d. che con la rettifica; di conseguenza la separazione finale viene fatta in un minor numero di stadi globali. La d. con rettifica è un'operazione comunissima nell'industria chimica; allorché è applicabile è quasi sempre il metodo più economico per frazionare le miscele liquide e gassose (previamente condensate).
![]()
![]()
Le grandi torri in cui si fa questa operazione costituiscono il simbolo delle raffinerie e dell'industria chimica in generale. Per il calcolo delle apparecchiature di d. e d. con rettifica sono necessari i dati termodinamici sulle miscele da trattare, e precisamente in primo luogo le relazioni di equilibrio liquido-vapore. Queste relazioni (espresse in una forma qualsiasi) dicono quale è la composizione della fase liquida e della fase gassosa fra loro in equilibrio in certe condizioni di temperatura e di pressione. Se sono poste in forma grafica si dicono diagrammi di stato liquido-gas. Tali relazioni sono essenzialmente sperimentali. è possibile in qualche caso farne un calcolo approssimato con certe formule, ed in casi rarissimi avere una legge generale da applicare. Si parla allora di miscele ideali, che obbediscono alla legge di Raoult. Per miscele a due componenti, che diremo A e B, questa si enuncia così: "la pressione parziale di ogni componente in una fase gassosa in equilibrio col suo liquido è data dal prodotto della tensione di vapore del componente (nelle stesse condizioni della miscela) moltiplicata per la sua frazione molare nella fase liquida". Esprimiamo questa legge in forma matematica. Anzitutto la frazione molare di un componente in una fase è data dal numero di moli di quel componente nella fase divisa per il numero di moli totali della fase. Nel nostro caso, se nA e nB sono il numero di moli di A e di B nella fase liquida questa sarà costituita di n = nA + nB moli. Pertanto le frazioni molari xA e xB di A e di B saranno date da:
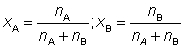
è evidente che xA + xB = 1, cioè la somma delle frazioni molari dei componenti di una miscela è l'unità. Se PA e PB sono le tensioni di vapore di A e B nelle condizioni della miscela (cioè le pressioni sotto le quali bollirebbero A e B puri, in un recipiente che contenga solo vapori di A e B puri nelle condizioni di temperatura della miscela e pressione pari a quella sopra la miscela) e pA e pB sono le loro pressioni parziali nel vapore sopra la miscela, sarà pA + pB = P pressione totale sopra la miscela. Allora la legge di Raoult si esprime così:
pA = xA PA; pB = xB PB
Dette yA e yB le frazioni molari del gas nella fase vapore (riservando xA e xB a quelle nella fase liquida), se anche la miscela gassosa è ideale (cosa che si verifica se vale la legge di Raoult) si ha che:
yA = pA/P; yB = pB/P
onde, sostituendo nelle relazioni viste sopra si trova:
yA = xA (PA/P); yB = xB (PB/P)
che legano la concentrazione di A e B nel vapore a quelle nel liquido, dato che le tensioni di vapore dei componenti sono constanti (a temperatura e pressione fissate). Se il componente A è più volatile di B si ha che PA > PB onde si può facilmente dimostrare che yA > xA mentre yB < xB. Si vede pertanto che la fase vapore è più ricca di A e più povera di B della fase liquida, come si è già detto. Per i calcoli si suole introdurre anche la volatilità relativa α che - se vale la legge di Raoult - si esprime come αAB= PA/PB, mentre nel caso generale vale:
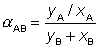
ulla volatilità relativa sono basati i calcoli delle colonne di d.; se si è in presenza di più componenti, si usano le volatilità relative di tutti meno uno rispetto a questo. I casi in cui la d. diventa impossibile (cioè non sortisce il suo scopo di frazionare la miscela) sono due: componenti a volatilità circa uguale o formazione di azeotropi (che è un caso particolare del precedente). Nel caso in cui le volatilità siano molto prossime all'unità, la separazione che si realizza ad ogni stadio di d. è molto bassa; al limite è nulla se le tensioni di vapore sono uguali e quindi α è unitario. Occorrerebbero allora un numero molto grande di stadi, al limite infinito. Si può allora modificare le volatilità relative introducendo altri composti, opportunamente scelti; naturalmente questi devono essere poi separati a loro volta onde il processo diviene più complicato. Questo caso sarà trattato più avanti. Una separazione difficile, che si fa per d. e rettifica con colonne molto alte è il frazionamento della miscela propano-propilene nei suoi componenti, i quali hanno volatilità circa uguale come si può vedere dai punti di ebollizione (propilene -47,0°C; propano -42,5°C). Essa richiede colonne con un centinaio di piatti. Nel caso in cui i componenti di una miscela formino un azeotropo si ha in quel punto volatilità relativa unitaria, e la miscela distilla e condensa invariata, onde non è possibile realizzare alcuna separazione mediante d. o rettifica. Anche in questi casi si ricorre ad artifici. Prima di passare alla trattazione dei vari tipi di d., occorre premettere che molto spesso essi vengono condotti per via grafica, utilizzando i diagrammi di stato liquido-vapore (di solito a P costante, cioè a pressione costante), cioè i cosiddetti diagrammi T-x (temperatura-composizione). Da questi diagrammi si ricavano per comodità di calcolo altri diagrammi, e precisamente i cosiddetti y-x, che danno la frazione molare di un componente in fase vapore (che è la y) in funzione di quella fase liquida (che è la x). Un esempio del passaggio da un diagramma T-x ad uno y-x è dimostrato in figura; sono pure illustrati i diagrammi nel caso di formazione di un azeotropo. Esaminiamo ora i casi più comuni di d. ║ D. semplice discontinua o batch: in questo caso - che è quello della d. per produrre la grappa citato sopra - una caldaia (o alambicco) viene caricata con un liquido e portata all'ebollizione. Il vapore che si sviluppa viene condensato facendolo passare in un serpentino raffreddato ad esempio con acqua. L'operazione procede fin che il distillato o il residuo hanno una certa composizione prefissata. Il frazionamento può essere reso più completo ripetendo più volte questa operazione; ogni volta si avrà però una quantità sempre minore di distillato. L'apparecchiatura necessaria è estremamente semplice. Per il calcolo si può usare la formula:
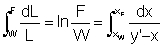
in cui: F = carica iniziale; L = quantità di residuo presente al tempo considerato; x = frazione molare nel residuo dello stesso relativa al componente che interessa; y' = frazione molare dello stesso gas in equilibrio col liquido; W = quantità finale di residuo; xF e xW = frazioni molari del complesso che interessa rispettivamente nell'alimentazione e nel residuo finale. La relazione, detta di Lord Rayleigh, può servire per determinare F, W, xF o xW (una delle quattro, quando sono note le altre tre). ║ D. semplice continua o flash distillation: in questo caso si tratta di un'operazione continua, come dice il nome. Il principio però è lo stesso del caso precedente. L'alimentazione passa in un pipe still, cioè in una tubazione riscaldata in una caldaia, indi viene inviata in un separatore. Da questo viene estratto in testa il gas, mentre dalla coda si ottiene il liquido. Si possono porre molte di queste apparecchiature in serie, con ricicli di liquido per ottenere una buona alimentazione. Il separatore gas-liquido è del tipo a ciclone. L'operazione può anche essere condotta sotto pressione ridotta; è usata abbastanza spesso per effettuare un primo "taglio" di una miscela in due frazioni, che vengono poi purificate separatamente, soprattutto nelle miscele di idrocarburi leggeri. ║ D. in corrente di vapore: si tratta ancora di una d. semplice, nella quale il calore necessario per l'evaporazione è fornito iniettando nella miscela vapore vivo. La temperatura alla quale avviene l'operazione può essere regolata in un ampio intervallo agendo sulla portata del vapore e sulla quantità di surriscaldamento con cui esso viene alimentato. Questa d. ha un grande vantaggio, che è quello di permettere la d. di componenti sensibili al calore ad una temperatura inferiore a quella che li danneggerebbe, ma senza avere gli inconvenienti che derivano dall'operare sotto vuoto. Infatti in questo caso la pressione totale dell'apparecchiatura è data da quella del vapore e dalla tensione di vapore dei componenti che distillano; si può quindi operare ad esempio ad un'atmosfera di pressione totale anche se essi hanno una tensione di vapore di qualche decina di millimetri di mercurio. Naturalmente si deve poi operare una separazione nella fase gassosa per ottenere i componenti voluti senza acqua. Ciò può essere fatto nella maggior parte dei casi in modo semplice; dato che questa d. si usa pressoché solo con componenti immiscibili con l'acqua allo stato liquido, basta condensare il distillato per realizzare con pochissima spesa la separazione. Questo tipo di d. è molto usato nell'industria petrolchimica, farmaceutica e alimentare.║ D. frazionata discontinua (o d. discontinua con riflusso o d. e rettifica discontinua con riflusso): basilarmente si opera come nella d. semplice discontinua, ma il condensato che si ottiene in testa alla colonna non viene estratto completamente, bensì una parte viene rinviata (liquida) in testa alla colonna di d. Essa scende in questa verso il basso, costituendo quello che si dice un riflusso (di liquido). La colonna è in generale riempita di corpi di forma opportuna o contiene altri dispositivi in modo da aumentare la superficie di contatto fra il liquido che scende ed i vapori che salgono. Si ha punto per punto un effetto di d. e uno di rettifica: lungo tutta la colonna si ha evaporazione dei componenti più volatili presenti nel liquido, mentre si ha condensazione nei prodotti più pesanti contenuti nel vapore. Di conseguenza il liquido che scende verso il basso si va arricchendo dei componenti più pesanti, mentre il gas che sale si va arricchendo in quelli più leggeri. La d. di questo tipo realizza quindi una buona separazione, tanto migliore quanto più alta è la colonna e quanto maggiore è la frazione di distillato che viene rimandata in colonna a costituire il riflusso. Si definisce un rapporto di riflusso R come il valore della frazione:
R = Lo/D
dove Lo è la portata di liquido di riflusso e D è la quantità di prodotto distillato che esce istantaneamente dalla colonna. ║ D. e rettifica continua (o d. e rettifica): si opera come nel caso precedente, salvo che l'alimentazione non viene fatta alla base della colonna ma ad una certa altezza (a 3/4 o a metà altezza circa); dalla testa è estratto in continuità il vapore che, condensato, costituisce in parte il prodotto leggero e in parte il riflusso; dalla coda è estratto invece, sempre in continuo, il prodotto pesante.
![]()
Guadagnare navigando! Acquisti prodotti e servizi.
Guadagnare acquistando online.
![]()
Di solito il riscaldamento del liquido presente in fondo alla colonna non è fatto direttamente ma in un'apparecchiatura a parte detta ribollitore. L'alimentazione può essere liquida, gassosa o mista; il tratto di colonna che sta sotto l'alimentazione viene detto di stripping o di esaurimento, mentre quello soprastante viene detto di rettifica o di arricchimento. Le apparecchiature in cui avviene questa operazione sono sempre delle torri, di diametro variabile da qualche decimetro a qualche metro e di altezza che può arrivare a 50 e più metri. Esse possono essere riempite di materiale adatto per il contatto gas-liquido oppure essere divise da piatti. Nel primo caso l'operazione avviene lungo tutta la colonna; nel secondo avviene solo sui piatti, onde è detta intermittente o a stadi. Esaminiamo un semplice metodo di calcolo per le colonne a piatti. Il risultato di questo calcolo, essendo assegnata una certa miscela di cui sono noti i dati termodinamici e la purezza voluta dei prodotti di testa e di coda è il numero di piatti teorici che essa deve avere. Da questo calcolo, tenendo conto che i piatti sono tali per cui non si realizza mai su di essi l'equilibrio completo fra gas e liquido, cioè dividendo per un fattore minore di uno detto efficienza del piatto, si ha il numero di piatti reali che deve avere la colonna. Dalla produttività che essa deve avere e da altre considerazioni si fissa quindi il suo diametro e la distanza fra i piatti, e quindi la sua altezza totale. Il risultato è quindi il dimensionamento della colonna dal punto di vista dell'ingegneria chimica. Nel calcolo esposto si fa riferimento ad una miscela a due componenti aventi volatilità abbastanza diversa in tutto il campo di composizioni che interessano; inoltre si faranno varie ipotesi semplificative. è superfluo ricordare che esistono altri metodi di calcolo; quello esposto fa uso di una costruzione grafica relativamente semplice ed è detto metodo grafico di Mc Cabe e Thiele. Questo metodo si basa su un bilancio materiale e termico di una sezione di colonna; a sua volta i bilanci sono basati sul principio di conservazione della massa e dell'entalpia. è evidente che quanto entra nella colonna (a regime) deve essere uguale a quanto ne esce, e quindi vale la relazione:
F = D + W
in cui F è l'alimentazione, D è il distillato e W è il residuo, tutti espressi in una certa unità (ad es. kg/ora oppure kmoli/ora). Lo stesso si può scrivere su un tronco di colonna: la massa di vapore che entra (dal basso) più la massa di liquido che entra (dall'alto) è uguale alla massa di vapore e di liquido che escono (dall'alto e dal basso rispettivamente). Gli stessi bilanci valgono ancora se riferiti alle entalpie di quanto entra e di quanto esce, tenuto conto di apporti termici per riscaldamenti o dispersioni termiche. In generale le colonne lavorano a temperatura vicina all'ambiente o sono coibentate, onde le perdite termiche si trascurano; inoltre si fa l'ipotesi semplificativa che i calori molari di evaporazione dei componenti siano fra loro uguali. Scrivendo il bilancio materiale ed entalpico fra una sezione della colonna posta sopra l'alimentazione e la testa della colonna, con opportune trasformazioni analitiche si ottiene l'equazione di una retta detta retta di lavoro del tronco superiore della colonna che è precisamente:
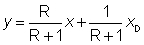
in cui R = Lo /D = rapporto di riflusso; xD = frazione molare del componente più volatile nel distillato; x ed y = coordinate del diagramma di stato x-y. Questa retta può essere riportata nel diagramma x-y per la miscela in esame, e sta fra la curva di equilibrio e la retta x = y (cioè la retta a 45°). Questa retta interseca la x = y nel punto x = xD ed interessa solo nel campo detto. Analogamente per il tronco inferiore della colonna gli stessi bilanci portano ad una retta, detta retta di lavoro del tronco inferiore della colonna, che ha equazione:
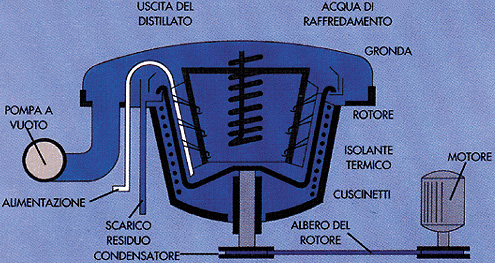
in cui i simboli hanno il valore noto, e inoltre: F = alimentazione; W = residuo di fondo colonna; xW = frazione molare del componente più leggero nel fondo colonna. Anche questa retta nel diagramma x-y sta, nel campo che interessa, fra la retta x = y e la curva di equilibrio; essa interseca la x = y nel punto di ascissa x = xW. Una volta fissato R (ad es. come tentativo) le due rette sono determinate, e si incontrano in un punto ben preciso. I due tratti di retta per il tronco superiore e inferiore costituiscono la linea di lavoro di tutta la colonna. Partendo da uno dei due estremi e tracciando successivamente dei segmenti orizzontali e verticali fra la linea di equilibrio e quella di lavoro ci si sposta fino a giungere od oltrepassare l'altro estremo (cioè si parte dal punto y = x = xD e si va fino ad y = x = xW o viceversa). Questi tratti hanno un significato ben preciso, ed esprimono delle condizioni di equilibrio e bilancio materiale fra piatto e piatto, di cui omettiamo la dimostrazione. Il numero di gradini così tracciato ha però un significato più preciso, in quanto è pari al numero di piatti teorici necessari per realizzare la separazione richiesta, cioè per avere un distillato avente frazione molare xD e un residuo avente frazione molare xW (sempre per il componente più leggero). Dai piatti teorici si passa quindi a quelli reali tenendo conto dell'efficienza. La costruzione serve anche a determinare su quale piatto va inviata l'alimentazione: è quello in corrispondenza al quale si ha l'intersezione fra le due rette di lavoro (del tronco superiore e di quello inferiore). Si può anche vedere che ponendo xD = 1 oppure xW = 0 si ottiene un numero infinito di piatti, il che significa che una separazione del tutto completa è impossibile (come si era già detto). Il rapporto di riflusso è molto importante per quanto riguarda il numero di piatti necessari; esso diminuisce infatti all'aumentare di R, ma contemporaneamente aumenta il consumo di calore per il ribollitore e di freddo per il condensatore, onde si deve trovare (a tentativi) un rapporto di riflusso opportuno. Molto spesso questo va da 2 a 5. Un altro metodo di calcolo spesso usato è il cosiddetto step by step o piatto a piatto. Esso ha il vantaggio di essere molto preciso e di fornire la composizione su tutti i piatti, dato che si parte dal piatto di testa (o di coda) e si calcola successivamente la composizione su tutti gli altri uno per uno ordinatamente. Richiede una mole notevole di calcolo, ma è molto adatto alla risoluzione mediante calcolatore elettronico, onde è di uso comune. Nel caso in cui si abbiano molti componenti, il calcolo diviene molto più complesso; si usano dei metodi approssimati. Questo caso è assai comune nella petrolchimica. Se necessario è possibile derivare da piatti a diverse altezze della colonna delle correnti laterali aventi una certa composizione. Ad esempio nella d. del petrolio grezzo, oltre al prodotto di testa e di coda della colonna si usa estrarre da 3 a 6 correnti laterali che forniscono miscele di idrocarburi aventi diverso punto di ebollizione. ║ D. azeotropica: è usata per miscele che presentano un azeotropo, e che non possono pertanto essere separate completamente. In questo caso, distillando e rettificando una miscela di composizione non azeotropica si ottengono due fasi: una composta da un composto praticamente puro, e l'altra composta dall'azeotropo. Il caso che si presenta spesse volte è che l'azeotropo, condensato, si smisceli in due fasi, cioè nei due componenti i quali sono poco solubili l'uno nell'altro. In questo caso è possibile separarli per decantazione: uno viene estratto tale e quale, mentre l'altro viene rinviato nella colonna (ed è poi estratto dal fondo di questa, se l'azeotropo è di massimo, caso più solito). Se il composto ottenuto per smiscelazione dell'azeotropo non è abbastanza puro, si può purificare ancora per d. e rettifica; si ottiene il composto puro e di nuovo l'azeotropo, che viene mandato al separatore di cui si è parlato prima. è questo il caso di molte miscele binarie, una delle più importanti è quella di acqua e n-butano. Se l'azeotropo non è smiscelabile, caso anche questo molto comune nella tecnica (ad es. acqua ed alcool etilico) non si può fare questa separazione, a meno di trovare un terzo componente che formi coi due un azeotropo ternario smiscelabile in due fasi che vengono poi trattate separatamente. è questo proprio il caso della miscela acqua-alcool etilico, che forma un azeotropo ternario con il benzene. ║ D. estrattiva: serve quando una miscela è composta di due (o più) componenti aventi circa la stessa volatilità, oppure che formano un azeotropo. Consiste nell'aggiungere alla miscela un terzo componente di natura tale da avere una volatilità molto diversa dai due, e tale da modificare la volatilità relativa dei due, di solito abbassando quella del meno volatile. Si differenzia dalla d. azeotropica con aggiunta di terzo componente per il fatto che non vi è formazione di azeotropo ternario, e che pertanto il componente aggiunto esce senz'altro in coda alla colonna, in miscela con un componente dal quale viene separato in una colonna a parte. Questa operazione è molto diffusa. Fra i casi più importanti ricordiamo la separazione del benzene dal cicloesano (aggiunta di fenolo), la separazione butene-butano (aggiunta di acetone e furfurolo), la separazione butadiene-butene (aggiunta acetone e furfurolo), la separazione isobutano-normal butene-1 e n-butano da n-butene-2 (aggiunta di furfurolo), la separazione isoprene-pentene (aggiunta di acetone), la separazione degli isoamileni, che servono per produrre isoprene, dalla frazione degli idrocarburi a 5 atomi di carbonio (aggiunta di acido solforico), la concentrazione dell'acido nitrico e cloridrico (aggiunta di acido solforico) e così via. Per quanto riguarda la scelta del terzo componente da aggiungere è possibile dare delle direttive generali, sulla base della polarità delle varie molecole; i dati definitivi per la progettazione devono però essere anche in questo caso sperimentali. L'apparecchiatura si calcola come una colonna di d., naturalmente a più componenti, dato che ve ne sono almeno tre. ║ D. molecolare: forma di d. applicabile a quelle sostanze che sono estremamente sensibili a un riscaldamento. Si opera sotto vuoto spinto fino a 0,001 mm Hg, benché nella pratica industriale le pressioni comuni siano comprese fra 0,003 e 0,03 mm Hg (cioè fra 3 e 30 micron di mercurio). è noto che il fenomeno dell'evaporazione di una sostanza avviene al livello molecolare come il distacco di molecole singole dalla massa liquida, allorché queste - trovandosi alla superficie del liquido - ricevono per effetto dell'agitazione termica un'energia cinetica sufficiente a vincere le forze di coesione che legano la molecola alla massa liquida. In tal modo un certo numero di molecole per unità di tempo passa nel gas che sovrasta il liquido, e si allontana da questo per diffusione. Non tutte le molecole però si comportano così; alcune, urtando contro molecole di gas, vengono "riflesse" verso la superficie liquida ed, entrando a contatto con questa, sono di nuovo "catturate" dal liquido. Questa riflessione, che in definitiva rende minore la velocità di evaporazione, può essere evitata in massima parte facendo avvenire la d. sotto un vuoto spinto. A 0,003 mm di mercurio il libero cammino medio delle molecole si approssima ad 1 cm, e gli urti detti divengono molto rari. Inoltre è ben noto il principio della parete fredda. Se un recipiente chiuso contiene una massa di liquido posta in una zona mantenuta calda mentre un'altra zona del recipiente è mantenuta fredda, pian piano il liquido evaporerà dalla zona calda e condenserà nella zona fredda fin che si sarà tutto raccolto in questa zona. Sullo stesso principio si basa la d. molecolare, solo che essa avviene alle pressioni bassissime dette. Inoltre perché essa avvenga bene si deve avere la zona fredda (o condensatore) molto vicino alla zona calda (o evaporatore). In pratica se il libero cammino medio delle molecole si approssima ad un centimetro, la distanza fra superficie di evaporazione e di condensazione può essere anche di 3÷4 cm senza che molte molecole evaporate ritornino al liquido da cui si sono staccate. L'apparecchiatura necessaria a realizzare la d. molecolare consiste essenzialmente in una superficie evaporante, riscaldata elettricamente, sulla quale si distende un sottile strato di liquido fermo o in moto laminare. Nelle immediate vicinanze vi è la superficie condensante opportunamente refrigerata. La costruzione può essere quanto mai varia secondo il tipo di lavoro; oggi però nella pratica industriale si sono affermate le apparecchiature centrifughe, in cui il liquido alimentato è disteso sulla superficie evaporante ad opera della forza centrifuga; la stessa serve per lo scarico del condensato e del residuo. Il tutto naturalmente è chiuso in un recipiente a tenuta in cui entra l'albero del rotore. Dato che la tenuta non è mai perfetta, il recipiente va naturalmente collegato sempre con una pompa a vuoto. Una di queste apparecchiature è illustrata in figura le dimensioni variano da pochi centimetri a circa 150 cm (di diametro); la velocità di rotazione è compresa fra 400 e 500 giri/minuto. Lo strato di liquido evaporante è compreso fra 0,005 e 0,1 mm di spessore. La capacità di d. va di solito da 100 a 1.000 litri/ora di alimentazione. Da un punto di vista impiantistico, questa d. si può considerare come una d. semplice continua. La d. quindi potrà essere più o meno spinta (cioè sarà possibile evaporare una quantità maggiore o minore del liquido alimentato) secondo il tempo di permanenza del liquido da evaporare sulla piastra riscaldata; a questo scopo si può effettuare una regolazione ad esempio variando il numero di giri del rotore. Naturalmente è anche possibile accoppiare più di queste apparecchiature, ponendole una in serie all'altra allorché si voglia una separazione tanto spinta da non essere ottenibile in un solo stadio. Questa d. è estremamente costosa; è quindi impiegata in quei campi in cui il costo del prodotto può ripagarla ampiamente, come ad esempio nella purificazione di medicinali e vitamine. Negli altri casi, allorché si ha un prodotto sensibile al calore, si ricorre alla d. in corrente di vapore mista, oppure alla d. sotto vuoto (cioè ad una d. e rettifica condotta a bassa pressione, ad es. a 100 ÷ 500 mm Hg). ║ D. distruttiva: d. che consiste in un forte riscaldamento fuori dal contatto dell'aria, al quale si sottopongono certe sostanze solide (legno, carboni fossili, scisti oleosi, ecc.) per liberare da essi sostanze volatili. Il termine distruttiva è usato perché spesso la liberazione di queste sostanze comporta una distruzione più o meno spinta del solido che le tratteneva, oppure addirittura una decomposizione termica di questo, con liberazione appunto di sostanze volatili. è questo il caso ad esempio della d. del legno; questo viene carbonizzato (cioè trasformato nel carbone di legna, costituito essenzialmente da carbonio) con liberazione di acqua, alcool metilico, acido acetico, ecc. La d. del carbon fossile è pure un'operazione quanto mai comune; si ottiene come residuo il coke (usato come combustibile od in metallurgia come riducente) ed il gas illuminante, usato come combustibile (il comune gas di città, che però di solito è miscelato con altri gas). La d. degli scisti oleosi porta all'ottenimento del cosiddetto olio di scisto, il quale per successivi trattamenti può essere trasformato ad esempio in olio lubrificante. La d. distruttiva è compiuta in storte. Per la descrizione delle apparecchiature usate per la d. e rettifica (V. DISTILLAZIONE, COLONNA DI).
Apparecchio per distillazione molecolare
![]()
![]()
Enciclopedia termini lemmi con iniziale a b c d e f g h i j k l m n o p q r s t u v w x y z
Storia Antica dizionario lemmi a b c d e f g h i j k l m n o p q r s t u v w x y z
Dizionario di Storia Moderna e Contemporanea a b c d e f g h i j k l m n o p q r s t u v w y z
Lemmi Storia Antica Lemmi Storia Moderna e Contemporanea
Dizionario Egizio Dizionario di storia antica e medievale Prima Seconda Terza Parte
Storia Antica e Medievale Storia Moderna e Contemporanea
Dizionario di matematica iniziale: a b c d e f g i k l m n o p q r s t u v z
Dizionario faunistico df1 df2 df3 df4 df5 df6 df7 df8 df9
Dizionario di botanica a b c d e f g h i l m n o p q r s t u v z
![]()
